QualTex Laboratories provides state-of-the art patient, donor, and biological testing services, screening millions of samples for international biotechnology customers every year.
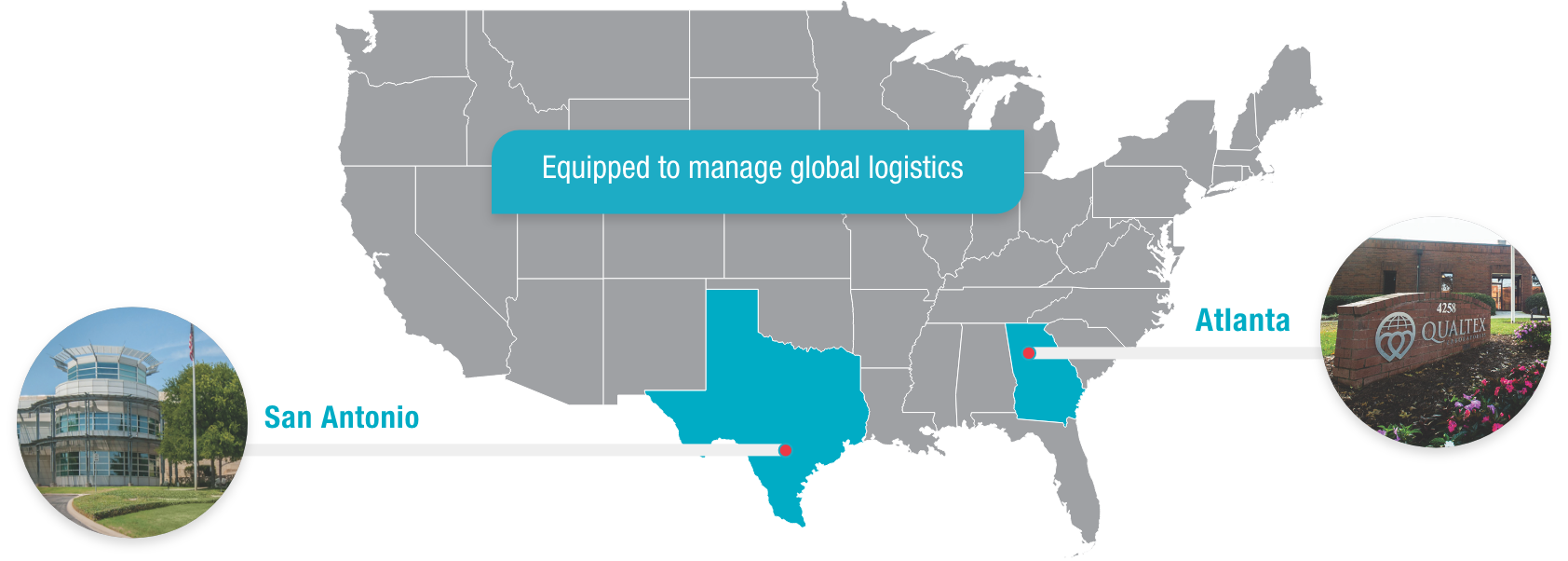
QualTex is one of the largest independent, nonprofit testing laboratories in the United States for blood and plasma products, with locations in San Antonio, Texas, and Norcross, Georgia.
Automation
The ACCELERATOR® a3600 Laboratory Automation System, now in use at QualTex facilities in Atlanta and San Antonio, is a powerful, flexible system designed to meet current and future needs for testing automation.
The ACCELERATOR® a3600 Laboratory Automation System, now in use at QualTex facilities in Atlanta and San Antonio, is a powerful, flexible system designed to meet current and future needs for testing automation.